Analysis of GUIDE-seq data to identify offtargets using GUIDEseq package
Lihua Julie Zhu
2020-07-28
Source:vignettes/GUIDEseqdemo.Rmd
GUIDEseqdemo.RmdReferences
Zhu LJ, Lawrence M, Gupta A, Pages H, Kucukural A, Garber M and Wolfe SA (2017). “GUIDEseq: a bioconductor package to analyze GUIDE-Seq datasets for CRISPR-Cas nucleases.” BMC Genomics, 18(1). http://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3746-y.
Main text of the article: Use cases
Additional File 1: Preprocessing steps to genereate alignment and umi files as input for GUIDEseq
Additional File 2: Installation and use of GUIDEseq (intended for new users of R and Bioconductor)
First load the GUIDEseq and annotation packages.
We are going to use a dataset generated from human samples, which has been included in the GUIDEseq package. To annotate the targets and off-targets, we need to load Human BSgenome package, Human Transcript and gene identifier mapping packages.
Note: It is critical to use the same version of the genome for sequence mapping (preprocessing) and for sequence analysis in GUIDEseq.
library(GUIDEseq)
## Loading required package: GenomicRanges## Loading required package: stats4## Loading required package: BiocGenerics## Loading required package: parallel##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, basename, cbind, colnames,
## dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
## grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
## rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
## union, unique, unsplit, which.max, which.min## Loading required package: S4Vectors##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:base':
##
## expand.grid## Loading required package: IRanges## Loading required package: GenomeInfoDblibrary(BSgenome.Hsapiens.UCSC.hg19)
## Loading required package: BSgenome## Loading required package: Biostrings## Loading required package: XVector##
## Attaching package: 'Biostrings'## The following object is masked from 'package:base':
##
## strsplit## Loading required package: rtracklayerlibrary(TxDb.Hsapiens.UCSC.hg19.knownGene)
## Loading required package: GenomicFeatures## Loading required package: AnnotationDbi## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.library(org.Hs.eg.db)
## Here are the commands to learn more about the GUIDEseq package and how to use the two workflow functions to perform GUIDE-seq data analysis for different types of CRISPR system and to compare different experiments.
help(GUIDEseqAnalysis) help(combineOfftargets) browseVignettes("GUIDEseq")
Example 1. Analysis of SpCas9 GUIDE-seq data
Although the analysis workflow function GUIDEseqAnalysis has more than 60 parameters for customized analysis, the majority of these parameters are pre-set for analyzing GUIDE-seq data from the most commonly used nuclease, SpCas9. Consequently when analyzing SpCas9 data only a small number of target-specific inputs are required from users.
There are four required inputs.
1) gRNA.file for specifying a file containing one or more gRNAs in fasta format;
2) alignment.file for specifying 1-2 files containing the sequence alignment in bam format;
3) umi.file for specifying 1-2 files containing UMI for each read;
4) BSgenomeName for specifying the BSgenome object containing the genome sequences
Please note that if outputDir is not specified, results will be automatically saved in a folder named according to the peak calling/merging/filtering criteria specified, e.g., gRNAmin80window20step20distance40 will be the default outputDir with the following code snippet.
The gRNA file can be created using a text editor or vi. Both alignment and umi files are generated in the preprocessing steps. The analysis results are saved as offTargetAnaysisOfPeaks.xls in the specified output directory.
umi.inputfile <- system.file("extdata", "UMI-HEK293_site4_chr13.txt", package = "GUIDEseq") alignment.inputfile <- system.file("extdata","bowtie2.HEK293_site4_chr13.sort.bam", package = "GUIDEseq") gRNA.file <- system.file("extdata","gRNA.fa", package = "GUIDEseq") guideSeqRes <- GUIDEseqAnalysis( alignment.inputfile = alignment.inputfile, umi.inputfile = umi.inputfile, gRNA.file = gRNA.file, orderOfftargetsBy = "peak_score", descending = TRUE, BSgenomeName = Hsapiens, min.reads = 80, n.cores.max = 4)
## Remove duplicate reads ...## Peak calling ...## computing coverage for plus strand ...## computing coverage for minus strand ...## call peaks ...## finding local max for chromosome: chr13## combine plus and minus peaks ...## keep peaks not in merged.gr but present in both peaks1 and peaks2## offtarget analysis ...## search for gRNAs for input file1...
## [1] "Scoring ..."
## >>> Finding all hits in sequence chr13+:27629413:27629420:chr13-:27629400:27629404 ...
## >>> DONE searching
## >>> Finding all hits in sequence chr13+:39262927:39262939:chr13-:39262918:39262920 ...
## >>> DONE searching
## finish off-target search in sequence 2
## finish off-target search in sequence 1
## finish feature vector building
## finish score calculation
## [1] "Done!"
## Done with offtarget search!
## Add gene and exon information to offTargets ....
## Extract PAM sequence and n.PAM.mismatch.## Please check output file in directory gRNAmin80window20step20distance40guideSeqRes$offTargets
## offTarget peak_score predicted_cleavage_score gRNA.name
## 2 chr13:+:39262912:39262934 947 1.2 HEK239_site4
## 1 chr13:-:27629404:27629426 690 3.2 HEK239_site4
## gRNAPlusPAM offTarget_sequence guideAlignment2OffTarget
## 2 GGCACTGCGGCTGGAGGTGGNGG AGCAGTGCGGCTAGAGGTGGTGG A...G.......A.......
## 1 GGCACTGCGGCTGGAGGTGGNGG GGCACTGGGGTTGGAGGTGGGGG .......G..T.........
## offTargetStrand mismatch.distance2PAM n.PAM.mismatch n.guide.mismatch
## 2 + 20,16,8 0 3
## 1 - 13,10 0 2
## PAM.sequence offTarget_Start offTarget_End chromosome
## 2 TGG 39262912 39262934 chr13
## 1 GGG 27629404 27629426 chr13Please note that the alignment file, umi file and gRNA file are included in the GUIDEseq package. To run the analysis for your own data, please specify the file paths for target sequence (gRNA.file), sequence alignment (alignment.inputfile) and UMI input files (umi.inputfile). The following code assumes that the input files are located in the ~/GUIDEseqSpCas9Input directory.
The analysis results offTargetsInPeakRegions.xls will be saved in the ~/guideSeqResults directory as specified by outputDir.
library(GUIDEseq) gRNA.file <- "~/GUIDEseqSpCas9Input/SpCas9gRNAexample.fa" alignment.inputfile <- c("~/GUIDEseqSpCas9Input/plusLibrary.sort.bam", "~/GUIDEseqSpCas9Input/minusLibrary.sort.bam") umi.inputfile <- c("~/GUIDEseqSpCas9Input/plusLibraryUMI.txt", "~/GUIDEseqSpCas9Input/minusLibraryUMI.txt") outputDir <- "~/guideSeqResults" guideSeqResults <- GUIDEseqAnalysis( alignment.inputfile = alignment.inputfile, umi.inputfile = umi.inputfile, gRNA.file = gRNA.file, BSgenomeName = Hsapiens, outputDir = outputDir)
By default, the predicted cleavage score is calculated using the weight matrix and scoring algorithm from the Zhang laboratory. To use the algorithm developed by the Root Laboratory, set the scoring.method = “CFDscore”.
Example 2: Annotate off-targets
With parameters txdb and orgAnn set to an organism-specific transcript object and gene ID mapping object respectively, off-target sites are annotated to indicate whether the offtargets overlap with gene bodies and whether they fall within an exon. Here is an example for SpCas9 GUIDE-seq data processing that annotates identified potential off-target sites with features from the human genome.
library(TxDb.Hsapiens.UCSC.hg19.knownGene) library(org.Hs.eg.db) outputDir <- "~/guideSeqResults" guideSeqResults <- GUIDEseqAnalysis( alignment.inputfile = alignment.inputfile, umi.inputfile = umi.inputfile, gRNA.file = gRNA.file, BSgenomeName = Hsapiens, txdb = TxDb.Hsapiens.UCSC.hg19.knownGene, orgAnn = org.Hs.egSYMBOL, outputDir = outputDir)
## Remove duplicate reads ...## Peak calling ...## computing coverage for plus strand ...## computing coverage for minus strand ...## call peaks ...## finding local max for chromosome: chr13## combine plus and minus peaks ...## keep peaks not in merged.gr but present in both peaks1 and peaks2## offtarget analysis ...## search for gRNAs for input file1...
## [1] "Scoring ..."
## >>> Finding all hits in sequence chr13+:27629413:27629420:chr13-:27629400:27629404 ...
## >>> DONE searching
## >>> Finding all hits in sequence chr13+:39262927:39262939:chr13-:39262918:39262920 ...
## >>> DONE searching
## >>> Finding all hits in sequence chr13+:41969948:41969968:chr13-:41969948:41969967 ...
## >>> DONE searching
## >>> Finding all hits in sequence chr13+:88900985:88901002:chr13-:88900981:88901000 ...
## >>> DONE searching
## finish off-target search in sequence 2
## finish off-target search in sequence 1
## finish feature vector building
## finish score calculation
## [1] "Done!"
## Done with offtarget search!
## Add gene and exon information to offTargets ....## 403 genes were dropped because they have exons located on both strands
## of the same reference sequence or on more than one reference sequence,
## so cannot be represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a
## GRangesList object, or use suppressMessages() to suppress this message.## Extract PAM sequence and n.PAM.mismatch.## Please check output file in directory ~/guideSeqResultsTo annotate off-targets in other genomes, set txdb and orgAnn accordingly. For example, set txdb to TxDb.Mmusculus.UCSC.mm10.knownGene and orgAnno to org.Mm.eg.db for mouse.
Example 3. Merge off-targets from multiple experiments to facilitate comparisons among different nuclease configurations or variants.
When evaluating novel nuclease treatment conditions or different Cas9 variants, it is common practice to include off-target analysis of standard platforms as controls. To aid in comparisons between different nucleases, off-targets identified by GUIDE-seq can be easily merged using the combineOfftargets function. Here is the example code to merge three experiments and generate a Venn diagram to depict the off-target overlaps among experiments.
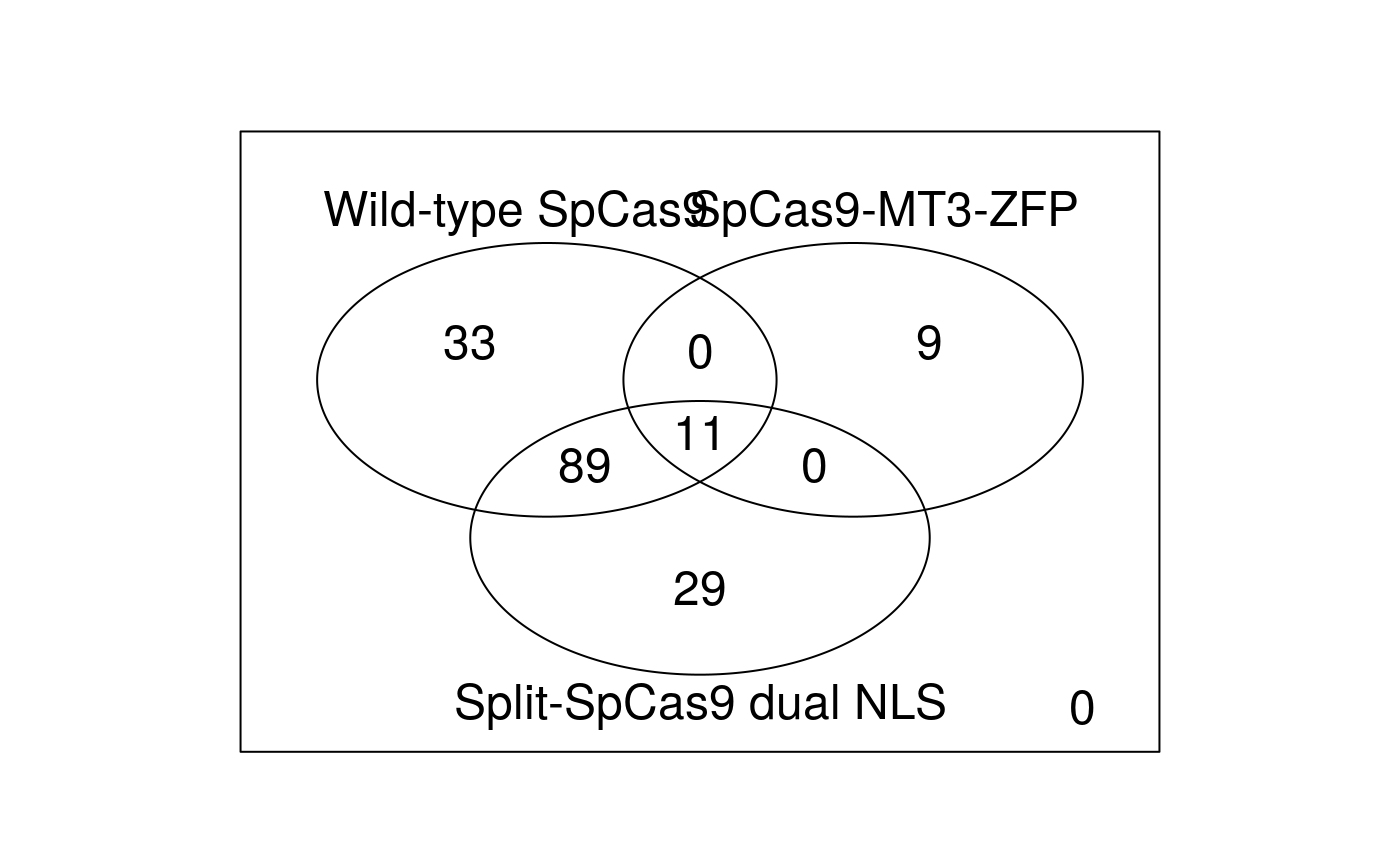
## offTarget predicted_cleavage_score gRNA.name
## 1 chr1:-:10926218:10926240 0.9 T2
## 2 chr1:-:178738721:178738743 0.1 T2
## 3 chr1:-:237195675:237195697 0.0 T2
## 4 chr1:-:51442186:51442208 0.1 T2
## 5 chr1:-:78245430:78245452 0.1 T2
## 6 chr1:+:11714529:11714551 0.8 T2
## gRNAPlusPAM offTarget_sequence guideAlignment2OffTarget
## 1 GACCCCCTCCACCCCGCCTCCGG CGAGCCCCCCACCCCGCCTCCGC CGAG...C............
## 2 GACCCCCTCCACCCCGCCTCCGG GGCCCTCTCCACTCCACCTCAGG .G...T......T..A....
## 3 GACCCCCTCCACCCCGCCTCCGG AACCCCACCCACCCCATCTCAGG A.....AC.......AT...
## 4 GACCCCCTCCACCCCGCCTCCGG GACCCCTCCCTCCCCACCTCAGG ......TC..T....A....
## 5 GACCCCCTCCACCCCGCCTCCGG TCTCCACCCCACCCCGCCCCTGG TCT..A.C..........C.
## 6 GACCCCCTCCACCCCGCCTCCGG GACCCGCCCCGCCCCGCCTCTGG .....G.C..G.........
## offTargetStrand mismatch.distance2PAM n.PAM.mismatch n.guide.mismatch
## 1 - 20,19,18,17,13 1 5
## 2 - 19,15,8,5 0 4
## 3 - 20,14,13,5,4 0 5
## 4 - 14,13,10,5 0 4
## 5 - 20,19,18,15,13,2 0 6
## 6 + 15,13,10 0 3
## PAM.sequence offTarget_Start offTarget_End chromosome
## 1 CGC 10926218 10926240 chr1
## 2 AGG 178738721 178738743 chr1
## 3 AGG 237195675 237195697 chr1
## 4 AGG 51442186 51442208 chr1
## 5 TGG 78245430 78245452 chr1
## 6 TGG 11714529 11714551 chr1
## Wild-type SpCas9.peak_score SpCas9-MT3-ZFP.peak_score
## 1 NA 3
## 2 9 NA
## 3 6 NA
## 4 26 NA
## 5 3 NA
## 6 10 NA
## Split-SpCas9 dual NLS.peak_score
## 1 NA
## 2 NA
## 3 9
## 4 14
## 5 11
## 6 7Please note that the above code assumes that each of the offtarget.folders contain the offTargetsInPeakRegions.xls file
If desired, combineOfftargets can be used to remove off-targets common among different gRNAs by setting remove.common to TRUE. Furthermore, if a control sample without nuclease is available, peaks present in the control sample can be removed from the gRNA samples by setting the control.sample.name.